Xiaoyan Li, Sophie Libkind, Nathaniel D. Osgood, Eric Redekopp and I have been creating software for modeling the spread of disease… with the help of category theory!
Lots of epidemiologists use “stock-flow diagrams” to describe ordinary differential equation (ODE) models of disease dynamics. We’ve created two tools to help them.
The first, called StockFlow.jl, is based on category theory and written in AlgebraicJulia, a framework for programming with categories that many people at or associated with Topos have been developing. The second, called ModelCollab, runs on web browsers and serves as a graphical user interface for StockFlow.jl.
Using ModelCollab requires no knowledge of Julia or category theory! This feature should be useful in “participatory modeling”, an approach where models are built with the help of diverse stakeholders. However, as we keep introducing new features in StockFlow.jl, it takes time to implement them in ModelCollab.
But what’s a stock-flow diagram, and what does our software let you do with them?
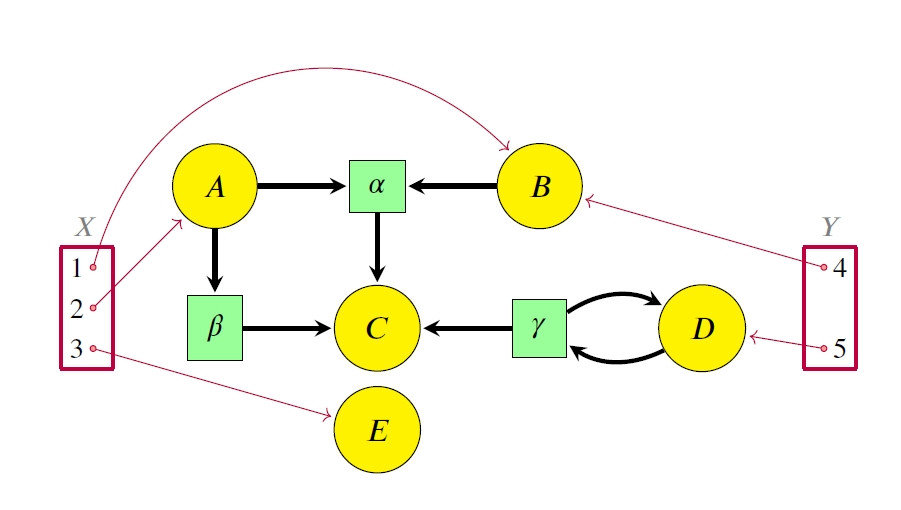
The picture here shows an example: a simple disease model where Susceptible people become Infective, then Recovered, then Susceptible again.
The boxes are “stocks” and the double-edged arrows are “flows”. There are also blue “links” from stocks to “variables”, and from stocks and variables to flows. This picture doesn’t show the formulas that say exactly how the variables depend on stocks, and how the flows depend on stocks and variables. So, this picture doesn’t show the whole thing. It’s really just what they call a “system structure diagram”: a stock-flow diagram missing the quantitative information that you need to get a system of ODEs from it. A stock-flow diagram, on the other hand, uniquely specifies a system of first-order ODEs.
Modelers often regard diagrams as an informal step toward a mathematically rigorous formulation of a model in terms of ODEs. However, we’ve shown that stock-flow diagrams have a precise mathematical syntax! They are objects in a category 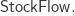 while “open” stock-flow diagrams, where things can flow in and out of the whole system, are horizontal 1-cells in a double category
while “open” stock-flow diagrams, where things can flow in and out of the whole system, are horizontal 1-cells in a double category 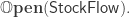 If you know category theory you can read a paper we wrote with Evan Patterson where we explain this:
If you know category theory you can read a paper we wrote with Evan Patterson where we explain this:
• John C. Baez, Xiaoyan Li, Sophie Libkind, Nathaniel D. Osgood and Evan Patterson, Compositional modeling with stock and flow diagrams. To appear in Proceedings of Applied Category Theory 2022.
If you don’t, we have a gentler paper for you:
• John C. Baez, Xiaoyan Li, Sophie Libkind, Nathaniel D. Osgood and Evan Redekopp, A categorical framework for modeling with stock and flow diagrams, to appear in Mathematics for Public Health, Springer, Berlin.
Why does it help to formalize the syntax of stock-flow diagrams using category theory? There are many reasons, but here are three:
1. Functorial Semantics
Our software lets modelers separate the syntax of stock and flow diagrams from their semantics: that is, the various uses to which these diagrams are put. Different choices of semantics are described via different functors. This idea, called “functorial semantics”, goes back to Lawvere and is popular in certain realms of theoretical computer science.
Besides the ODE semantics, we have implemented functors that turn stock-flow diagrams into other widely used diagrams: “system structure diagrams”, which I already explained, and “causal loop diagrams”. It doesn’t really matter much here, but a causal loop diagram ignores the distinction between stocks, flows and variables, lumps them all together, and has arrows saying what affects what:
These other forms of semantics capture purely qualitative features of stock and flow models. In the future, people can implement still more forms of semantics, like stochastic differential equation models!
So, instead of a single monolithic model, we have something much more flexible.
2. Composition
ModelCollab provides a structured way to build complex stock-flow diagrams from small reusable pieces. These pieces are open stock-flow diagrams, and sticking together amounts to composing them.
ModelCollab lets users save these diagrams and retrieve them for reuse as parts of various larger models. Since ModelCollab can run on multiple web browsers, it lets members of a modeling team compose models collaboratively. This is a big advance on current systems, which are not optimized for collaborative work.
This picture shows two small stock-flow diagrams being composed in ModelCollab:
Some of the underlying math here was developed in earlier work using categories and epidemiological modeling, which was also done by people at Topos and their collaborators:
• Sophie Libkind, Andrew Baas, Micah Halter, Evan Patterson and James P. Fairbanks, An algebraic framework for structured epidemic modelling, Philosophical Transactions of the Royal Society A 380 (2022), 20210309.
3. Stratification
Our software also allows users to “stratify” models: that is, refine them by subdividing a single population (stock) into several smaller populations with distinct features. For example, you might take a disease model and break each stock into different age groups.
In contrast to the global changes commonly required to stratify stock-flow diagrams, our software lets users build a stratified diagram as a “pullback” of simpler diagrams, which can be saved for reuse. Pullbacks are a concept from category theory, and here we are using pullbacks in the category whose objects are system structure diagrams. Remember, these are like stock and flow diagrams, but lacking the quantitative information describing the rates of flows. After a system structure diagram has been constructed, this information can be added to obtain a stock and flow diagram.
This picture shows two different models stratified in two different ways, creating four larger models. I won’t try to really explain this here. But at least you can get a tiny glimpse of how complicated these models get. They get a lot bigger! That’s why we need software based on good math to deal with them efficiently.
References
[AJ] AlgebraicJulia: Bringing compositionality to technical computing.
[B1] John C. Baez, Xiaoyan Li, Sophie Libkind, Nathaniel D. Osgood and Evan Patterson, Compositional modeling with stock and flow diagrams. To appear in Proceedings of Applied Category Theory 2022.
[B2] John C. Baez, Xiaoyan Li, Sophie Libkind, Nathaniel D. Osgood and Eric Redekopp, A categorical framework for modeling with stock and flow diagrams, to appear in Mathematics for Public Health, Springer, Berlin.
[H] P. S. Hovmand, Community Based System Dynamics, Springer, Berlin, 2014.
[L] Sophie Libkind, Andrew Baas, Micah Halter, Evan Patterson and James P. Fairbanks, An algebraic framework for structured epidemic modelling, Philosophical Transactions of the Royal Society A 380 (2022), 20210309.
[MC] ModelCollab: A web-based application for collaborating on simulation models in real-time using Firebase.
[SF] Stockflow.jl.



 Posted by John Baez
Posted by John Baez 








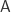 is a category with finite
is a category with finite  is a
is a 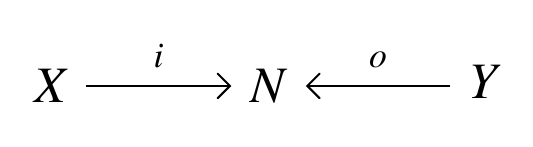
 called a decoration. Then there is a symmetric monoidal category with
called a decoration. Then there is a symmetric monoidal category with -decorated cospans as morphisms.
-decorated cospans as morphisms. of our cospan with some extra structure: we do this by choosing an element of some set
of our cospan with some extra structure: we do this by choosing an element of some set 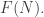 But in practice, we often want to decorate
But in practice, we often want to decorate 
 is not just a category but a 2-category: it has categories as objects, functors as morphisms, but also natural transformations as 2-morphisms. The natural notion of ‘something like a functor’ from a category to a 2-category is called a
is not just a category but a 2-category: it has categories as objects, functors as morphisms, but also natural transformations as 2-morphisms. The natural notion of ‘something like a functor’ from a category to a 2-category is called a  and for any
and for any  we want
we want 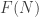 to be the set, or category, of finite graphs having
to be the set, or category, of finite graphs having 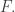 And instead of just giving you a symmetric monoidal category, this theorem gives you a symmetric monoidal double category.
And instead of just giving you a symmetric monoidal category, this theorem gives you a symmetric monoidal double category. in which
in which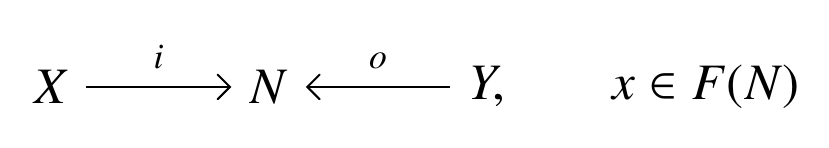
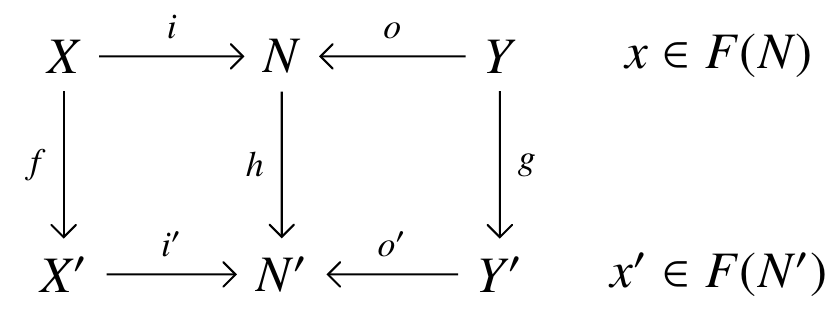
 the map of decorations.
the map of decorations. appears in an adjunction
appears in an adjunction  ? (By the way, all monoidal categories in this post will be strict, so I’m going to drop that adjective for conciseness.)
? (By the way, all monoidal categories in this post will be strict, so I’m going to drop that adjective for conciseness.) and single object
and single object 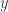 , a set of arrows from
, a set of arrows from 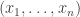 to
to 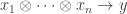 ; thus we can now have multiple generating objects in the domains of generating morphisms, but not the codomains.
; thus we can now have multiple generating objects in the domains of generating morphisms, but not the codomains.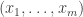 and
and  . I’d be tempted to call this a “polygraph”, since it also naturally generates a polycategory. But other folks got there first and called it a “tensor scheme” and also a “pre-net”. In the latter case, the objects are called “places” and the morphisms “transitions”. But whatever we call it, it allows us to generate free symmetric monoidal categories in which the domains and codomains of generating morphisms can both be arbitrary tensor products of generating objects. For those who like fancy higher-categorical machinery, it’s the notion of
. I’d be tempted to call this a “polygraph”, since it also naturally generates a polycategory. But other folks got there first and called it a “tensor scheme” and also a “pre-net”. In the latter case, the objects are called “places” and the morphisms “transitions”. But whatever we call it, it allows us to generate free symmetric monoidal categories in which the domains and codomains of generating morphisms can both be arbitrary tensor products of generating objects. For those who like fancy higher-categorical machinery, it’s the notion of  and a transition
and a transition  , while a second pre-net
, while a second pre-net  has three places
has three places  and a transition
and a transition 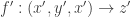 . Once we generate a symmetric monoidal category, then
. Once we generate a symmetric monoidal category, then 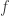 can be composed with a symmetry
can be composed with a symmetry 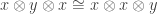 and similarly for
and similarly for 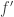 ; so the symmetric monoidal categories generated by
; so the symmetric monoidal categories generated by  and
and 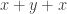 respectively, which in a commutative monoid are equal (both are
respectively, which in a commutative monoid are equal (both are  ). So the corresponding Petri nets of
). So the corresponding Petri nets of 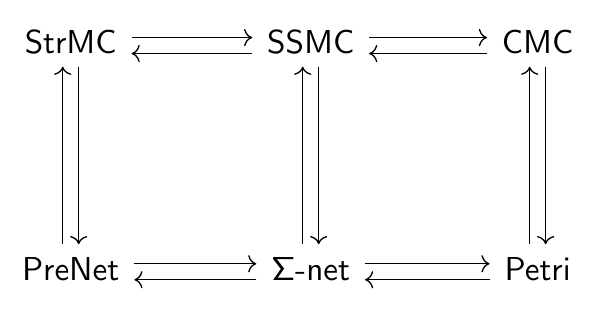
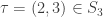 , yielding a new morphism
, yielding a new morphism  which we can take to be the image of
which we can take to be the image of  uniquely determines where we have to send all these permutation images.
uniquely determines where we have to send all these permutation images. , a groupoid
, a groupoid 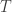 , and a discrete opfibration
, and a discrete opfibration  , where
, where 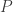 denotes the free-symmetric-strict-monoidal-category functor
denotes the free-symmetric-strict-monoidal-category functor  . Such a discrete opfibration is the same as a functor
. Such a discrete opfibration is the same as a functor  , and the objects of
, and the objects of  are the finite sequences of elements of
are the finite sequences of elements of  by the transposition
by the transposition  that switches the first two entries. We get another transition
that switches the first two entries. We get another transition  with the same domain and codomain as
with the same domain and codomain as  , then when we generate a free symmetric monoidal category from our Σ-net, the corresponding morphism
, then when we generate a free symmetric monoidal category from our Σ-net, the corresponding morphism  will have the property that when we compose it with the symmetry morphism
will have the property that when we compose it with the symmetry morphism  we get back
we get back 
